

08 Apr First article published using the new Fusion MS
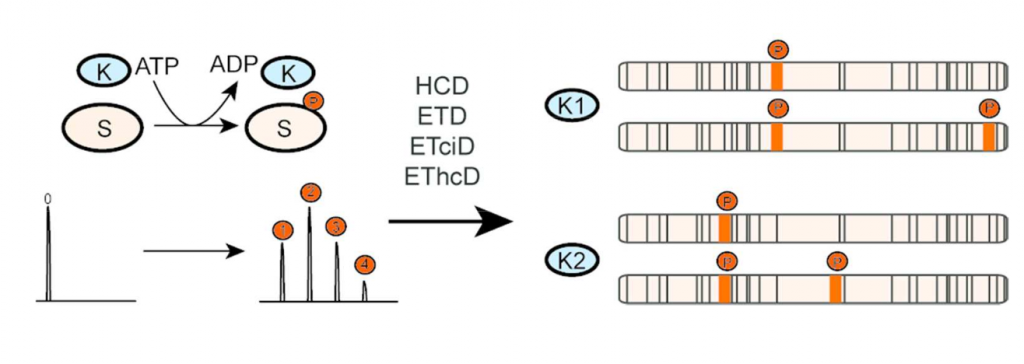
Top-down analysis of intact proteins by mass spectrometry provides an ideal platform for comprehensive proteoform characterization, in particular, for the identification and localization of post-translational modifications (PTM) co-occurring on a protein. One of the main bottlenecks in top-down proteomics is insufficient protein sequence coverage caused by incomplete protein fragmentation. Based on previous work on peptides, increasing sequence coverage and PTM localization by combining sequential ETD and HCD fragmentation in a single fragmentation event, we hypothesized that protein sequence coverage and phospho-proteoform characterization could be equally improved by this new dual fragmentation method termed EThcD, recently been made available on the Orbitrap Fusion. Here, we systematically benchmark the performance of several (hybrid) fragmentation methods for intact protein analysis on an Orbitrap Fusion, using as a model system a 17.5 kDa N-terminal fragment of the mitotic regulator Bora. During cell division Bora becomes multiply phosphorylated by a variety of cell cycle kinases, including Aurora A and Plk1, albeit at distinctive sites. Here, we monitor the phosphorylation of Bora by Aurora A and Plk1, analyzing the generated distinctive phospho-proteoforms by top-down fragmentation. We show that EThcD and ETciD on a Fusion are feasible and capable of providing richer fragmentation spectra compared to HCD or ETD alone, increasing protein sequence coverage, and thereby facilitating phosphosite localization and the determination of kinase specific phosphorylation sites in these phospho-proteoforms. Data are available via ProteomeXchange with identifier PXD001845.
![]()
Sorry, the comment form is closed at this time.